The Role of UCL’s Cell and Gene Therapy Experts in Shaping the Field’s Future. By Nhi Pham and Nidhi Rege
In recent years, the number of cell and gene therapies (CGTs) approved for clinical trials has skyrocketed. These ground-breaking treatments leverage engineered cells or genetic material to target and cure diseases - even those long considered incurable. CGTs have massive implications for the future of our healthcare systems, heralding a monumental shift towards personalised medicine. Despite their tremendous promise, many biological, financial, and ethical challenges must be overcome as these technologies transition from bench to bedside.
University College London is no stranger to this rapidly advancing field, with the institution being hailed as the superpower of Europe (1). Along with its partner NHS Trust, UCL actively participates in a remarkable 59% of the UK's advanced therapy clinical trials, taking a pivotal role in their progression.
However, as the scope of opportunity grows, so do its ethical debates. Twenty UCL academics and experts in CGTs were interviewed and surveyed for their perspectives on these expanding technologies. The ethical concerns discussed primarily revolved around:
- Fears of the field’s rapid evolution
- Navigating regulatory challenges
- Accessibility and reimbursement issue
- The role of eugenics
- Widening healthcare disparities
- Use of CGTs in utero
Fear of the field’s rapid evolution
Despite the potential offered by CGTs, they are still in their infancy. In 2018, the global scientific community was stunned by He Jiankui's announcement that he had assisted in producing the world's first genetically edited babies. This premature endeavour was met with widespread outrage from the scientific and medical community, and Chinese regulators were quick to respond with strong condemnation and a three-year prison sentence for "illegal medical practice” (2). This controversy not only triggered a fear of gene editing among the general public but also highlighted the challenges posed by disruptive technologies that outpace existing regulations and ethical practices in place
In response, the World Health Organization (WHO) established the Expert Advisory Committee on Developing Global Standards for Governance and Oversight of Human Genome Editing. This committee played a pivotal role in leading global efforts to regulate genome editing. In 2021, they released the first global recommendations to establish human genome editing as a public health tool emphasising safety, effectiveness, and ethics. This includes three reports: a position paper, summarising their findings; a governance framework on human genome editing; and the committee’s overall recommendations (3–5). Conclusively, their findings reinforce the notion that we are not yet adequately prepared to conduct heritable germline editing in humans, (i.e., the ‘designer baby’ hype). As such, according to a 2020 CRISPR Journal survey, heritable germline editing is not explicitly permitted in any country around the world (6).
Navigating Regulatory Challenges
The aftermath of the groundbreaking event marked by the first human gene editing, combined with its potential ethical and safety challenges, highlights the critical need for establishing robust government frameworks for CGT. For this reason, the process of bringing a CGT product to market is a long, rigorous journey involving multiple steps designed to ensure safety and efficacy. In Europe, CGT falls into the category of Advanced Therapy Medicinal Products (ATMPs) (7). ATMPs receive regularly updated guidelines detailing expectations on quality, pre-clinical, and clinical evidence (8).
To ensure safety and consistency between batches, a substantial submission of paperwork and data must be reviewed before the first-in-human phase 1/2 clinical trial begins. Pre-clinical data begins with the proof-of-concept studies (discovery phase), followed by in vitro data and, finally, in vivo studies using appropriate animal models. The final set of documentation required is a detailed clinical trial protocol, including the number of participants, inclusion and exclusion criteria, dosing strategy, and detailed primary and secondary endpoints to ensure a clear purpose.
For ATMPs, there is no separate Phase 1 trial involving healthy participants. Instead, only patients diagnosed with the target disease are allowed to participate due to the greater risks associated with ATMPs compared with traditional drugs (10). As the priority in Phase 1/2 trials is safety rather than efficacy, doses start lower than what would be expected to have a therapeutic effect. Only once the data suggest this to be safe is the dose increased. After successful Phase 1/2 data and constant engagement with regulatory bodies, a larger Phase 3 trial is initiated, often administered more widely in several hospitals, sometimes worldwide (11).
Due to the lengthy process of recruiting sufficient participants to meet the stringent inclusion/exclusion criteria and the complexity associated with manufacturing and administering ATMPs, clinical trials take many years to complete. This also includes lengthy mandatory long-term monitoring to ensure the therapy's long-term effects are known, as far as possible. According to Prof. Bobby Gaspard, CEO of Orchard Therapeutics “All patients who have been treated in our trials will continue to be followed for 15 years”, adding that, for his company’s gene therapy product, the focus goes beyond the clinical realm and extends to the molecular level. As novel therapeutics, ATMPs face much stricter scrutiny than typical small molecule drugs, with stringent post-authorisation requirements to ensure that any long-term side effects are detected.
Ultimately, the development of an ATMP from concept to clinic takes more than 10 years, hopefully reassuring to members of the general public who feared the unregulated administration of therapies. Of course, it is always difficult to predict how well results from animal studies will translate into humans, and there are always risks. This is a new branch of medicine, and there is still a lot that remains to be learned about the human body, but regulatory agencies are dedicated to creating and enforcing protocols aimed at minimising risk in the development process.
Accessibility and Reimbursement issues
Given the scale of the financial injection needed to bring a therapy through the complex approval process described, drug companies set a considerably high price to recoup their investments. Furthermore, as patient populations that can benefit from these treatments are very small, profits generated from a limited number of treatments must compensate for the entire drug development costs. For this reason, cost is at the centre of the ethical debate. The expensive price tags associated with these emerging treatments are a major barrier to the accessibility of much of the global patient population.
Although current regulations thoroughly scrutinise drug pricing, gene therapy costs remain largely unregulated and are determined on a case-by-case basis. Under a traditional reimbursement system, the cost of cell and gene therapy would often entail a substantial up-front payment, that could reach up to several hundred thousand to a few million per patient.
Despite Professor Amit Nathwani’s initial dream of developing a gene therapy accessible in the developing world, this vision remains unrealized due to the lack of affordability for these cutting-edge treatments. Roctavian was developed for severe haemophilia A based on the research led by Professor Nathwani and his team at UCL and St. Jude Children’s Research Hospital. Expected to secure FDA approval as early as this spring, Roctavian carries a projected list price of $2.9 million per use and a price per vial that equates to about $900,000 in Germany (12).
Even healthcare systems in high-income countries struggle to withstand the substantial up-front costs of these emerging therapies. For this reason, negotiations with healthcare payers can be long, which may lead to rejections. In 2021, US-based Bluebird Bio was forced to withdraw Zynteglo, their approved gene therapy for beta-thalassemia, from the European market after failing to reach a price (15). The challenges in securing satisfactory reimbursement terms indicate a lack of support from EU payers, who failed to recognise the innovation and long-term value of these products for patients. This serves as a discouraging precedent for other ATMP developers anticipating launches in Europe. In addition, therapy withdrawals may occur if there is an insufficient return on investment, essentially bankrupting the company. The unfortunate part is that there are patients with high levels of unmet needs who would have benefited from these promising treatments.
The Role of Eugenics
Eugenics is defined by its founder Francis Galton as “the science that improves stock”(16). This definition in its historical context has classist and racist connotations, with whiteness and wealth being associated with intellect and success. As the field of CGTs has evolved, the role of eugenics has been concerning for many spectators. The line between improving features and characteristics for medical benefit or to fit into prejudiced societal biases has been incredibly blurred(17). Over half of UCL academics are concerned over the increasing role of eugenics in the field. A particular academic believes that regulation is not a concept unique to CGTs and the first hurdle is that society must come to an agreement over what is ethically acceptable. A good place to start, perhaps, would be to decide upon a boundary to demarcate therapeutic and cosmetic benefits as well as societal expectation.
How early is too early?
CGT can be used in adults and children, but the idea of using these treatments before the child is even born, i.e. intrauterine, has excited many researchers across the field (18). But how early is too early?
38% of UCL academics surveyed believe that it is never too early to use these therapies.
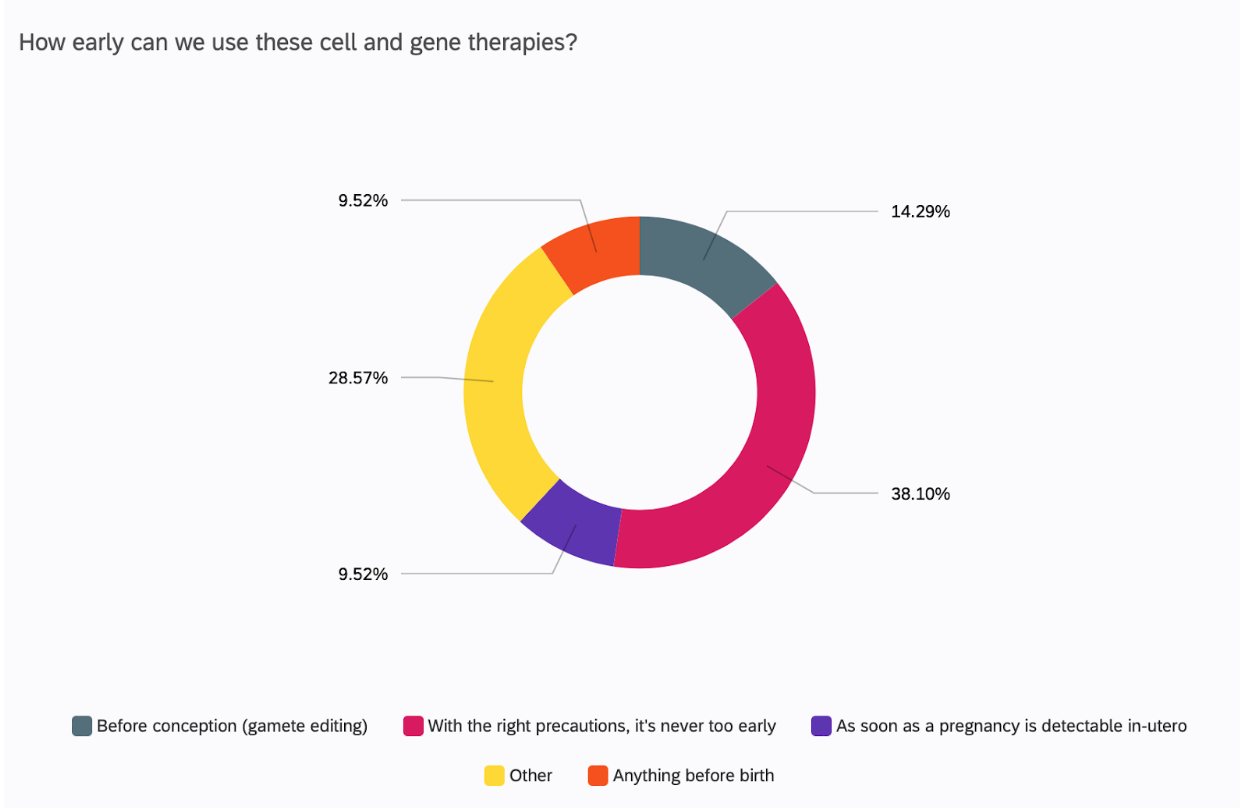
However, the technical and ethical reality behind using these therapies as soon as possible is not so optimistic. One respondent discussed editing in utero, but dismissed it briskly after, citing the stagnant ethical discourse where society is undecided regarding editing in utero. So, whilst UCL academics at large are supportive of pre-birth use of gene therapy, they themselves admit that the world is not. A US study revealed that in-utero therapies are actually less accepted than therapies in adults and children, with the largest fear being of unexpected medical issues down the line(19).
Another respondent had a somewhat different outlook on when gene therapies should be used. They believed that it all circles back to the infamous debate on when life begins. This particular person felt like it was unnecessary to 'waste' the time and money on correcting mutations in a foetus when you could simply abort and have another child. Or, in the case of in-vitro fertilisation, select an embryo without that genetic aberration.
Our Thoughts
It is a no-brainer that the future of medicine and biomedical engineering depends heavily on CGTs. The field is supported by the enthusiasm and passion of researchers such as the ones interviewed for this article, at prestigious institutions like UCL. However, it could be expected that these individuals would believe so strongly in these therapies. After all, there must be a good reason why they have poured their life’s work into this study.
The general worry is that if too enthusiastic, there will be a forced imposition of these drugs onto the public. Not so much medically, but financially, as the NHS is taxpayer funded and many UK Government grants in Research and Development are essentially funded by the citizens. For now, that does not seem to be an issue as more people believe that scientists and physicians should make these decisions instead of policy-makers themselves(20).
Besides public opinion, is the UK National Health Service pragmatically prepared to use these therapies? As discussed previously, these therapies are incredibly expensive. Is it sustainable for the taxpayer-funded service to keep purchasing these drugs when other treatment plans may cost less and have acceptable patient outcomes? Within the NHS, there have been concerns about whether pharmacies and drug storages are prepared for the logistical issues of dispensing these drugs (e.g., storage, transport, and adequate training). If it is accepted that CGTs are the future, research must be done into the practical issues they face. Concurrently, whilst public support is in line with that of academics, it must be monitored to ensure that biomedical studies in the field remain accountable to the public they serve.
Article written by Nhi Pham and Nidhi Rege
References:
Cover photo credit: Sangharsh Lohakare/ Unsplash
- CELL, GENE AND REGENERATIVE THERAPIES AT UCL AND NHS PARTNER TRUSTS World leaders in the translation of advanced therapies Great Ormond Street Hospital for Children NHS Foundation Trust [Internet]. 2018 [cited 2023 Dec 8]. Available from: https://www.ucl.ac.uk/translational-research/sites/translational-research/files/cgr_therapies_report.pdf
- Cyranoski D. What CRISPR-baby prison sentences mean for research. Nature [Internet]. 2020 Jan 3;577(7789):154–5. Available from: https://www.nature.com/articles/d41586-020-00001-y
- WHO. Human genome editing: position paper [Internet]. www.who.int. 2021. Available from: https://www.who.int/publications/i/item/9789240030404
- WHO. Human genome editing: a framework for governance [Internet]. www.who.int. 2021. Available from: https://www.who.int/publications/i/item/9789240030060
- WHO. Human genome editing: recommendations [Internet]. www.who.int. 2021. Available from: https://www.who.int/publications/i/item/9789240030381
- Baylis F, Darnovsky M, Hasson K, Krahn TM. Human Germline and Heritable Genome Editing: The Global Policy Landscape. The CRISPR Journal [Internet]. 2020 Oct 20;3(5):365–77. Available from: https://www.liebertpub.com/doi/10.1089/crispr.2020.0082
- European Union. COMMISSION DIRECTIVE 2009/120/EC [Internet]. 2009. Available from: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2009:242:0003:0012:EN:PDF
- EMA. Guidelines relevant for advanced therapy medicinal products | European Medicines Agency [Internet]. www.ema.europa.eu. [cited 2023 Dec 8]. Available from: https://www.ema.europa.eu/en/human-regulatory-overview/advanced-therapy-medicinal-products-overview/guidelines-relevant-advanced-therapy-medicinal-products
- Leo L. BioMarin prices hemophilia gene therapy at nearly 29,000 euros per vial in Germany. Reuters [Internet]. 2023 Nov 28 [cited 2023 Dec 8]; Available from: https://www.reuters.com/business/healthcare-pharmaceuticals/biomarin-prices-hemophilia-gene-therapy-nearly-29000-euros-per-vial-germany-2023-11-28/#:~:text=Roctavian
- Pizevska M, Kaeda J, Fritsche E, Elazaly H, Reinke P, Amini L. Advanced Therapy Medicinal Products’ Translation in Europe: A Developers’ Perspective. Frontiers in Medicine [Internet]. 2022 Feb 3 [cited 2022 Sep 19];9:757647. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8851388/
- Detela G, Lodge A. EU Regulatory Pathways for ATMPs: Standard, Accelerated and Adaptive Pathways to Marketing Authorisation. Molecular Therapy - Methods & Clinical Development [Internet]. 2019 Jun [cited 2019 Jun 24];13:205–32. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6378853/pdf/main.pdf
- Reed J. Libmeldy: World’s “most expensive” drug recommended for NHS use. BBC News [Internet]. 2022 Feb 4 [cited 2022 Apr 18]; Available from: https://www.bbc.co.uk/news/health-60245738
- McPhillips D. FDA approves $3.5 million treatment for hemophilia, now the most expensive drug in the world [Internet]. CNN. 2022 [cited 2023 May 21]. Available from: https://edition.cnn.com/2022/11/23/health/hemophilia-drug-hemgenix/index.html
- Helmore E. Gene therapy at $3.5m a dose approved for US adults with hemophilia B. The Guardian [Internet]. 2022 Nov 23 [cited 2023 Feb 27]; Available from: https://www.theguardian.com/business/2022/nov/23/hemgenix-hemophilia-b-fda-most-expensive-drug
- Pagliarulo N. Bluebird to withdraw gene therapy from Germany after dispute over price. BioPharma Dive [Internet]. 2021 Apr 20 [cited 2021 Apr 20]; Available from: https://www.biopharmadive.com/news/bluebird-withdraw-zynteglo-germany-price/598689/
- AGAR N. Why We Should Defend Gene Editing as Eugenics. Cambridge Quarterly of Healthcare Ethics. 2019 Jan 20;28(1):9–19.
- HARRIS J. IS GENE THERAPY A FORM OF EUGENICS? Bioethics. 1993;7(2–3):178–87.
- Peddi NC, Ramesh HM, Gude SS, Gude SS, Vuppalapati S. Intrauterine Fetal Gene Therapy: Is That the Future and Is That Future Now? Cureus [Internet]. 2022 Feb 23 [cited 2023 Nov 16];14(2). Available from: /pmc/articles/PMC8951626/
- Vasquez-Loarte TC, Lucas TL, Harris-Wai J, Bowen DJ. Beliefs and Values About Gene Therapy and In-Utero Gene Editing in Patients with Hemophilia and Their Relatives. Patient. 2020 Oct 1;13(5):633–42.
- Aiyegbusi OL, Macpherson K, Elston L, Myles S, Washington J, Sungum N, et al. Patient and public perspectives on cell and gene therapies: a systematic review. Nat Commun [Internet]. 2020 Dec 1 [cited 2023 Nov 16];11(1). Available from: /pmc/articles/PMC7722871/
- Stoner N. Are UK hospital pharmacy departments ready for the rise of gene therapy medicinal products? Expert Opin Biol Ther [Internet]. 2018 Aug 3 [cited 2023 Nov 16];18(8):837–40. Available from: https://www.tandfonline.com/doi/abs/10.1080/14712598.2018.1495192